Publicaciones
Publicaciones seleccionadas
2026
2026
-
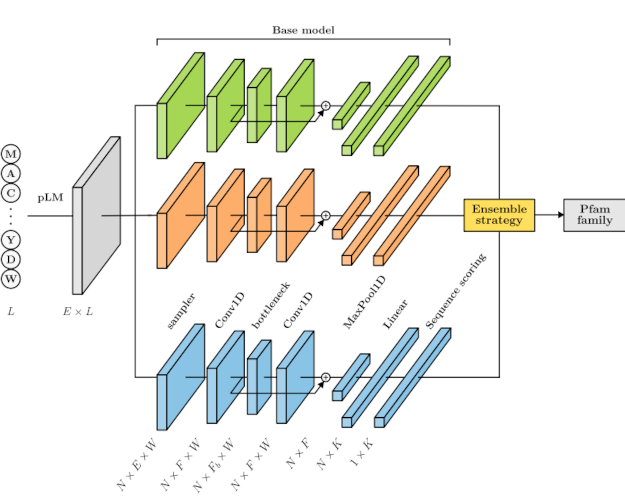 ET-Pfam: ensemble transfer learning for protein family predictionSofia A Duarte, Rosario Vitale, Sofia Escudero, and 4 more authorsBioinformatics, Apr 2026
ET-Pfam: ensemble transfer learning for protein family predictionSofia A Duarte, Rosario Vitale, Sofia Escudero, and 4 more authorsBioinformatics, Apr 2026Due to the rapid growth of sequence generation, which has surpassed the expert curators ability to manually review and annotate them, the computational annotation of proteins remains a significant challenge in bioinformatics nowadays. The Pfam database contains a large collection of proteins that are annotated with domain families through profile Hidden Markov models (pHMMs). Using the aligned sequences of a curated family, one HMM is trained independently for each family, missing the opportunity of learning patterns across families, i.e. from a complete view of all the dataset. As an alternative, some deep learning (DL) models have been recently proposed, nevertheless with simple representations of the inputs and moderate improvements in performance. In this work, we present ET-Pfam, a novel approach based on transfer learning and ensembles of multiple DL classifiers to predict functional families in the Pfam database. Several base DL models are first trained using learned representations from protein large language models. Then, the base models are integrated using classical ensemble strategies and novel voting approaches by learning weights for each model and for each Pfam family. Results demonstrate that the proposed ET-Pfam method can consistently diminish error rates compared to individual DL models, boosting prediction performance. Among the novel ensemble strategies presented here, the learned weights by family voting achieved the best performance, with the lowest error rate (7.00%), significantly surpassing the best individual base model error (12.91%) and competitors of the state-of-the-art. Data and source code are available at https://github.com/sinc-lab/ET-Pfam.
-
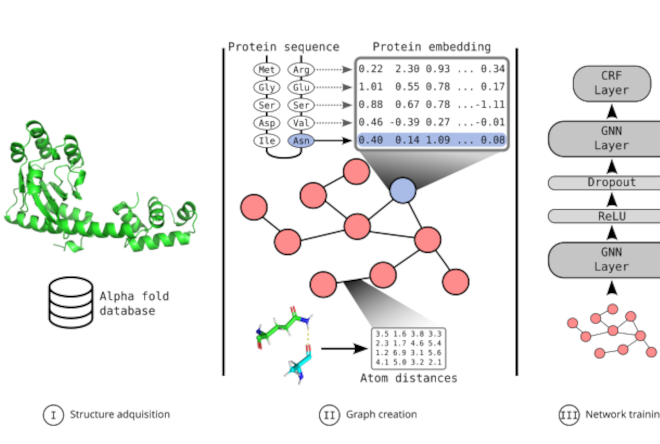 GNN2Pfam: Integrating protein sequence and structure with graph neural networks for Pfam domain annotationEmilio Fenoy, Leandro A Bugnon, Rosario Vitale, and 3 more authorsJournal of Structural Biology, Feb 2026
GNN2Pfam: Integrating protein sequence and structure with graph neural networks for Pfam domain annotationEmilio Fenoy, Leandro A Bugnon, Rosario Vitale, and 3 more authorsJournal of Structural Biology, Feb 2026The challenge of establishing the relationship between protein sequences and their function cannot yet be considered completely solved. State-of-the-art annotation of Pfam domains is based on hidden Markov models (HMMs) built from hand-crafted sequence alignments. However, while this approach has been highly successful during the last decades since its proposal, there is yet a very large number of proteins that remain unannotated because there is no possible alignment to already known and functionally characterized sequences, or HMM fails to discriminate between similar domains. Adding structural information using deep and graph neural networks (GNNs) presents an opportunity to build upon existing models in those more challenging cases. GNNs excel at capturing complex relationships in data and can learn a model that shares information across all existing families, thus being able to generalize Pfam domain predictions to novel sequences. In this protocol we propose GNN2Pfam, an end-to-end GNN-based method for Pfam family domain annotation. Our strategy allows one single model to be trained for all species and families. This novel proposal uses the protein 3D structure together with a sequence representation obtained from a large pre-trained model. The GNN2Pfam method is based on a graph derived from amino acid interactions in the 3D structure, learning both sequential and structural features from this representation. Experiments show that the proposed GNN-based model can clearly outperform the HMM state-of-the-art predictive performance in Pfam domains annotations. These results suggest that GNN models can be the key component of future protein annotation tools. Data and source code are available at https://github.com/efenoy/GNN2Pfam.
- emb2dis: a novel protein disorder prediction tool based on ResNets, dilated convolutions & protein language modelsSofia A Duarte, Mahta Mehdiabadi, Leandro A Bugnon, and 5 more authorsBioRxiv (under review), Mar 2026
Intrinsically disordered proteins (IDPs) play an important role in a wide range of biological functions and are linked to several diseases. Due to technical difficulties and the high cost of experimental determination of disorder in proteins, combined with the exponential increase of unannotated protein sequences, the development of computational methods for disorder prediction became an active area of research in the last few decades. Here we present emb2dis, a deep learning model that uses protein language models (pLMs) to predict disorder from sequence. The emb2dis tool is a pre-trained model that receives as input a protein sequence, calculates its pLM embedding and passes it to a deep learning model. In contrast to existing approaches, emb2dis integrates informative sequence representations with a novel architecture that combines residual networks (ResNets) and dilated convolutions.
2025
2025
- Big team science reveals promises and limitations of machine learning efforts to model physiological markers of affective experienceNicholas A Coles, Bartosz Perz, Maciej Behnke, and 53 more authorsRoyal Society Open Science, Jun 2025
Researchers are increasingly using machine learning to study physiological markers of emotion. We evaluated the promises and limitations of this approach via a big team science competition. Twelve teams competed to predict self-reported affective experiences using a multi-modal set of peripheral nervous system measures. Models were trained and tested in multiple ways: with data divided by participants, targeted emotion, inductions, and time. In 100% of tests, teams outperformed baseline models that made random predictions. In 46% of tests, teams also outperformed baseline models that relied on the simple average of ratings from training datasets. More notably, results uncovered a methodological challenge: multiplicative constraints on generalizability. Inferences about the accuracy and theoretical implications of machine learning efforts depended not only on their architecture, but also how they were trained, tested, and evaluated. For example, some teams performed better when tested on observations from the same (vs. different) subjects seen during training. Such results could be interpreted as evidence against claims of universality. However, such conclusions would be premature because other teams exhibited the opposite pattern. Taken together, results illustrate how big team science can be leveraged to understand the promises and limitations of machine learning methods in affective science and beyond.
-
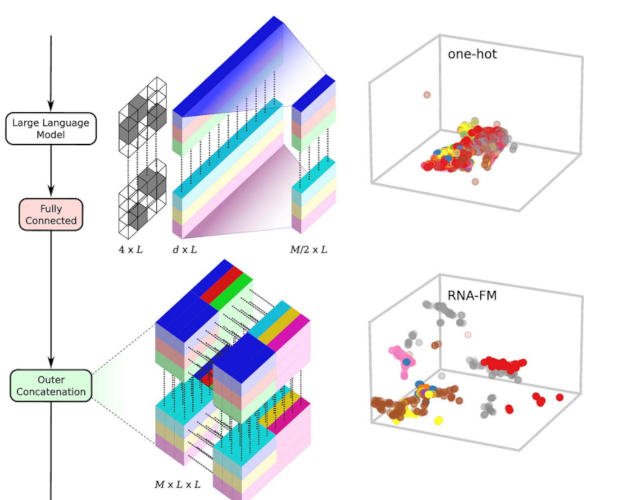 Comprehensive benchmarking of large language models for RNA secondary structure predictionLuciano I Zablocki, Leandro A Bugnon, Matias Gerard, and 3 more authorsBriefings in Bioinformatics, Jun 2025
Comprehensive benchmarking of large language models for RNA secondary structure predictionLuciano I Zablocki, Leandro A Bugnon, Matias Gerard, and 3 more authorsBriefings in Bioinformatics, Jun 2025In recent years, inspired by the success of large language models (LLMs) for DNA and proteins, several LLMs for RNA have also been developed. These models take massive RNA datasets as inputs and learn, in a self-supervised way, how to represent each RNA base with a semantically rich numerical vector. This is done under the hypothesis that obtaining high-quality RNA representations can enhance data-costly downstream tasks, such as the fundamental RNA secondary structure prediction problem. However, existing RNA-LLM have not been evaluated for this task in a unified experimental setup. Since they are pretrained models, assessment of their generalization capabilities on new structures is a crucial aspect. Nonetheless, this has been just partially addressed in literature. In this work we present a comprehensive experimental and comparative analysis of pretrained RNA-LLM that have been recently proposed. We evaluate the use of these representations for the secondary structure prediction task with a common deep learning architecture. The RNA-LLM were assessed with increasing generalization difficulty on benchmark datasets. Results showed that two LLMs clearly outperform the other models, and revealed significant challenges for generalization in low-homology scenarios. Moreover, in this study we provide curated benchmark datasets of increasing complexity and a unified experimental setup for this scientific endeavor. Source code and curated benchmark datasets with increasing complexity are available in the repository: https://github.com/sinc-lab/rna-llm-folding/
2024
2024
-
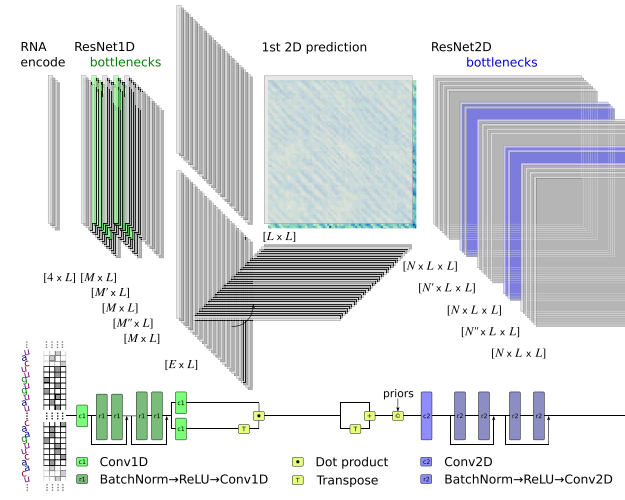 sincFold: end-to-end learning of short- and long-range interactions in RNA secondary structureLeandro A Bugnon, Leandro Di Persia, Matias Gerard, and 7 more authorsBriefings in Bioinformatics, Jun 2024
sincFold: end-to-end learning of short- and long-range interactions in RNA secondary structureLeandro A Bugnon, Leandro Di Persia, Matias Gerard, and 7 more authorsBriefings in Bioinformatics, Jun 2024Coding and noncoding RNA molecules participate in many important biological processes. Noncoding RNAs fold into well-defined secondary structures to exert their functions. However, the computational prediction of the secondary structure from a raw RNA sequence is a long-standing unsolved problem, which after decades of almost unchanged performance has now re-emerged due to deep learning. Traditional RNA secondary structure prediction algorithms have been mostly based on thermodynamic models and dynamic programming for free energy minimization. More recently deep learning methods have shown competitive performance compared with the classical ones, but there is still a wide margin for improvement.In this work we present sincFold, an end-to-end deep learning approach, that predicts the nucleotides contact matrix using only the RNA sequence as input. The model is based on 1D and 2D residual neural networks that can learn short- and long-range interaction patterns. We show that structures can be accurately predicted with minimal physical assumptions. Extensive experiments were conducted on several benchmark datasets, considering sequence homology and cross-family validation. sincFold was compared with classical methods and recent deep learning models, showing that it can outperform the state-of-the-art methods.
-
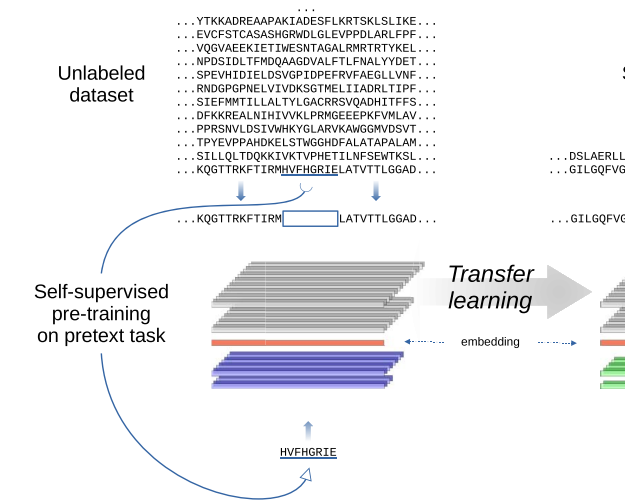 Evaluating large language models for annotating proteinsRosario Vitale, Leandro A Bugnon, Emilio Luis Fenoy, and 2 more authorsBriefings in Bioinformatics, May 2024
Evaluating large language models for annotating proteinsRosario Vitale, Leandro A Bugnon, Emilio Luis Fenoy, and 2 more authorsBriefings in Bioinformatics, May 2024In UniProtKB, up to date, there are more than 251 million proteins deposited. However, only 0.25\% have been annotated with one of the more than 15000 possible Pfam family domains. The current annotation protocol integrates knowledge from manually curated family domains, obtained using sequence alignments and hidden Markov models. This approach has been successful for automatically growing the Pfam annotations, however at a low rate in comparison to protein discovery. Just a few years ago, deep learning models were proposed for automatic Pfam annotation. However, these models demand a considerable amount of training data, which can be a challenge with poorly populated families. To address this issue, we propose and evaluate here a novel protocol based on transfer learningṪhis requires the use of protein large language models (LLMs), trained with self-supervision on big unnanotated datasets in order to obtain sequence embeddings. Then, the embeddings can be used with supervised learning on a small and annotated dataset for a specialized task. In this protocol we have evaluated several cutting-edge protein LLMs together with machine learning architectures to improve the actual prediction of protein domain annotations. Results are significatively better than state-of-the-art for protein families classification, reducing the prediction error by an impressive 60\% compared to standard methods. We explain how LLMs embeddings can be used for protein annotation in a concrete and easy way, and provide the pipeline in a github repo. Full source code and data are available at https://github.com/sinc-lab/llm4pfam
2023
2023
-
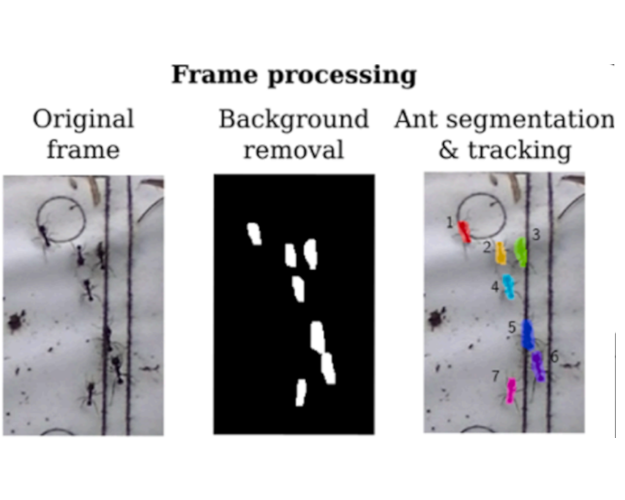 AntTracker: A low-cost and efficient computer vision approach to research leaf-cutter ants behaviorJulian Alberto Sabattini, Francisco Sturniolo, Martín Bollazzi, and 1 more authorSmart Agricultural Technology, May 2023
AntTracker: A low-cost and efficient computer vision approach to research leaf-cutter ants behaviorJulian Alberto Sabattini, Francisco Sturniolo, Martín Bollazzi, and 1 more authorSmart Agricultural Technology, May 2023Leaf-cutter ants play a crucial role in agroecosystems, and understanding their behavior is key to developing effective damage control strategies. While several tracking solutions exist for ants in controlled environments or on aerial images, accurately measuring ant behavior in the wild remains a challenge. In this work, we propose a three-stage processing pipeline that segments individual ants, tracks their movement, and classifies whether they are carrying a leaf using a convolutional neural network. The output of the pipeline includes a timestamped record of the activity on the trail, accounting for parameters such as ant velocity, size and if it is going from or to the nest. We use the recently developed portable device AntVideoRecord to register video of a selected ant trail. To validate our approach, we collected a labeled dataset and evaluated each stage using standard metrics, achieving a median F2 score of 83% for segmentation, MOTA of 97% for tracking and F1 of 82% for detecting ants carrying a leaf. We then carried out a larger use case in the wild, demonstrating the effectiveness of our approach in capturing the intricate behaviors of leaf-cutter ants. We believe our method has the potential to inform the development of more effective ant damage control strategies in agroecosystems.
-
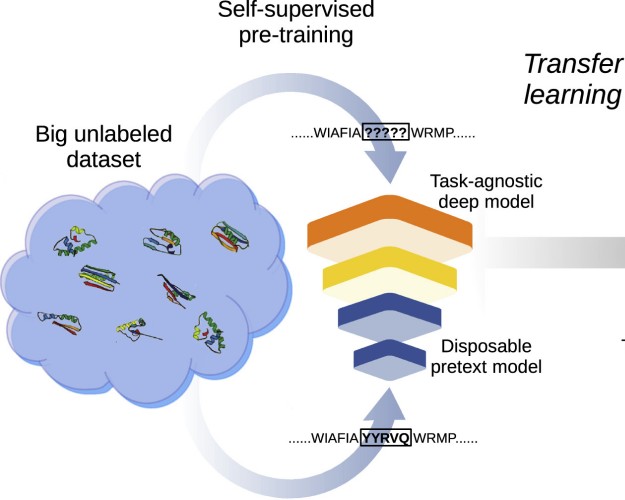 Transfer learning: The key to functionally annotate the protein universeL.A. Bugnon, E. Fenoy, A Edera, and 3 more authorsPatterns, May 2023
Transfer learning: The key to functionally annotate the protein universeL.A. Bugnon, E. Fenoy, A Edera, and 3 more authorsPatterns, May 2023The automatic annotation of the protein universe is still an unresolved challenge. Today, there are 229,149,489 entries in the UniProtKB database, but only 0.25% of them have been functionally annotated. This manual process integrates knowledge from the protein families database Pfam, annotating family domains using sequence alignments and hidden Markov models. This approach has grown the Pfam annotations at a low rate in the last years. Recently, deep learning models appeared with the capability of learning evolutionary patterns from unaligned protein sequences. However, this requires large-scale data, while many families contain just a few sequences. Here, we contend this limitation can be overcome by transfer learning, exploiting the full potential of self-supervised learning on large unannotated data and then supervised learning on a small labeled dataset. We show results where errors in protein family prediction can be reduced by 55% with respect to standard methods.
2022
2022
-
 Secondary structure prediction of long noncoding RNA: review and experimental comparison of existing approachesL. A. Bugnon, A. Edera, S. Prochetto, and 10 more authorsBriefing in Bioinformatics, May 2022
Secondary structure prediction of long noncoding RNA: review and experimental comparison of existing approachesL. A. Bugnon, A. Edera, S. Prochetto, and 10 more authorsBriefing in Bioinformatics, May 2022In contrast to messenger RNAs, the function of the wide range of existing long noncoding RNAs (lncRNAs) largely depends on their structure, which determines interactions with partner molecules. Thus, the determination or prediction of the secondary structure of lncRNAs is critical to uncover their function. Classical approaches for predicting RNA secondary structure have been based on dynamic programming and thermodynamic calculations. In the last 4 years, a growing number of machine learning (ML)-based models, including deep learning (DL), have achieved breakthrough performance in structure prediction of biomolecules such as proteins and have outperformed classical methods in short transcripts folding. Nevertheless, the accurate prediction for lncRNA still remains far from being effectively solved. Notably, the myriad of new proposals has not been systematically and experimentally evaluated. In this work, we compare the performance of the classical methods as well as the most recently proposed approaches for secondary structure prediction of RNA sequences using a unified and consistent experimental setup. We use the publicly available structural profiles for 3023 yeast RNA sequences, and a novel benchmark of well-characterized lncRNA structures from different species. Moreover, we propose a novel metric to assess the predictive performance of methods, exclusively based on the chemical probing data commonly used for profiling RNA structures, avoiding any potential bias incorporated by computational predictions when using dot-bracket references. Our results provide a comprehensive comparative assessment of existing methodologies, and a novel and public benchmark resource to aid in the development and comparison of future approaches. Full source code and benchmark datasets are available at: https://github.com/sinc-lab/lncRNA-folding
-
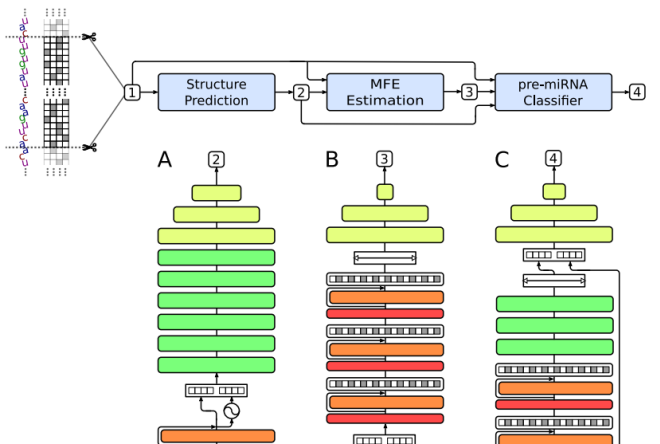 miRe2e: a full end-to-end deep model based on transformers for prediction of pre-miRNAsJ. Raad, L. A. Bugnon, D. H. Milone, and 1 more authorBioinformatics, May 2022
miRe2e: a full end-to-end deep model based on transformers for prediction of pre-miRNAsJ. Raad, L. A. Bugnon, D. H. Milone, and 1 more authorBioinformatics, May 2022MicroRNAs (miRNAs) are small RNA sequences with key roles in the regulation of gene expression at post-transcriptional level in different species. Accurate prediction of novel miRNAs is needed due to their importance in many biological processes and their associations with complicated diseases in humans. Many machine learning approaches were proposed in the last decade for this purpose, but requiring handcrafted features extraction to identify possible de novo miRNAs. More recently, the emergence of deep learning (DL) has allowed the automatic feature extraction, learning relevant representations by themselves. However, the state-of-art deep models require complex pre-processing of the input sequences and prediction of their secondary structure to reach an acceptable performance. In this work, we present miRe2e, the first full end-to-end DL model for pre-miRNA prediction. This model is based on Transformers, a neural architecture that uses attention mechanisms to infer global dependencies between inputs and outputs. It is capable of receiving the raw genome-wide data as input, without any pre-processing nor feature engineering. After a training stage with known pre-miRNAs, hairpin and non-harpin sequences, it can identify all the pre-miRNA sequences within a genome. The model has been validated through several experimental setups using the human genome, and it was compared with state-of-the-art algorithms obtaining 10 times better performance. Webdemo available at https://sinc.unl.edu.ar/web-demo/miRe2e/ and source code available for download at https://github.com/sinc-lab/miRe2e.
2021
2021
-
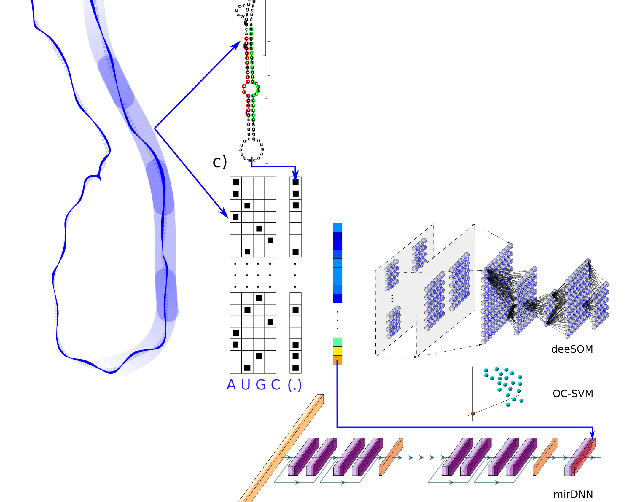 Deep Learning for the discovery of new pre-miRNAs: Helping the fight against COVID-19L. A. Bugnon, J. Raad, G.A. Merino, and 4 more authorsMachine Learning with Applications, May 2021
Deep Learning for the discovery of new pre-miRNAs: Helping the fight against COVID-19L. A. Bugnon, J. Raad, G.A. Merino, and 4 more authorsMachine Learning with Applications, May 2021The Severe Acute Respiratory Syndrome-Coronavirus 2 (SARS-CoV-2) has been recently found responsible for the pandemic outbreak of a novel coronavirus disease (COVID-19). In this work, a novel approach based on deep learning is proposed for identifying precursors of small active RNA molecules named microRNA (miRNA) in the genome of the novel coronavirus. Viral miRNA-like molecules have shown to modulate the host transcriptome during the infection progression, thus their identification is crucial for helping the diagnosis or medical treatment of the disease. The existence of the mature miRNAs derived from computationally predicted miRNA precursors (pre-miRNAs) in the novel coronavirus was validated with small RNA-seq data from SARS-CoV-2-infected human cells. The results demonstrate that computational models can provide accurate and useful predictions of pre-miRNAs in the SARS-CoV-2 genome, underscoring the relevance of machine learning in the response to a global sanitary emergency. Moreover, the interpretability of our model shed light on the molecular mechanisms underlying the viral infection, thus contributing to the fight against the COVID-19 pandemic and the fast development of new treatments. Our study shows how recent advances in machine learning can be used, effectively, in response to public health emergencies. The approach developed in this work could be of great help in future similar emergencies to accelerate the understanding of the singularities of any viral agent and for the development of novel therapies. Data and source code available at: https://sourceforge.net/projects/sourcesinc/files/aicovid
-
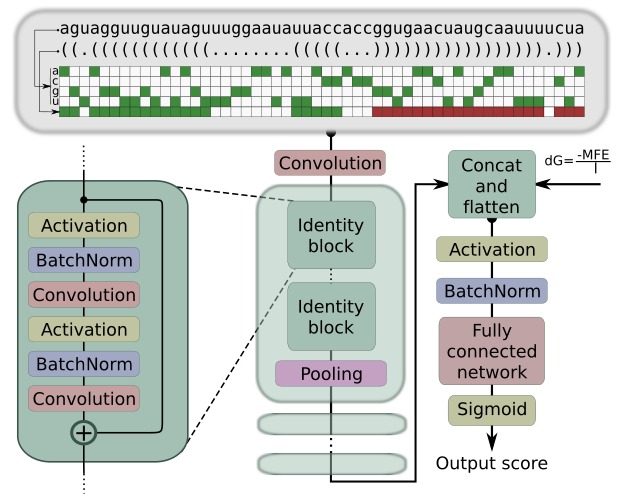 High precision in microRNA prediction: a novel genome-wide approach with convolutional deep residual networksC. Yones, J. Raad, L. A. Bugnon, and 2 more authorsComputers in Biology and Medicine, May 2021
High precision in microRNA prediction: a novel genome-wide approach with convolutional deep residual networksC. Yones, J. Raad, L. A. Bugnon, and 2 more authorsComputers in Biology and Medicine, May 2021MicroRNAs (miRNAs) are small non-coding RNAs that have a key role in the regulation of gene expression. The importance of miRNAs is widely acknowledged by the community nowadays and computational methods are needed for the precise prediction of novel candidates to miRNA. This task can be done by searching homologous with sequence alignment tools, but results are restricted to sequences that are very similar to the known miRNA precursors (pre-miRNAs). Besides, a very important property of pre-miRNAs, their secondary structure, is not taken into account by these methods. To fill this gap, many machine learning approaches were proposed in the last years. However, the methods are generally tested in very controlled conditions. If these methods were used under real conditions, the false positives increase and the precisions fall quite below those published. This work provides a novel approach for dealing with the computational prediction of pre-miRNAs: a convolutional deep residual neural network (mirDNN). This model was tested with several genomes of animals and plants, the full-genomes, achieving a precision up to 5 times larger than other approaches at the same recall rates. Furthermore, a novel validation methodology was used to ensure that the performance reported in this study can be effectively achieved when using mirDNN in novel species. To provide fast an easy access to mirDNN, a web demo is available at http://sinc.unl.edu.ar/web-demo/mirdnn/ . The demo can process FASTA files with multiple sequences to calculate the prediction scores and generates the nucleotide importance plots.
2020
2020
-
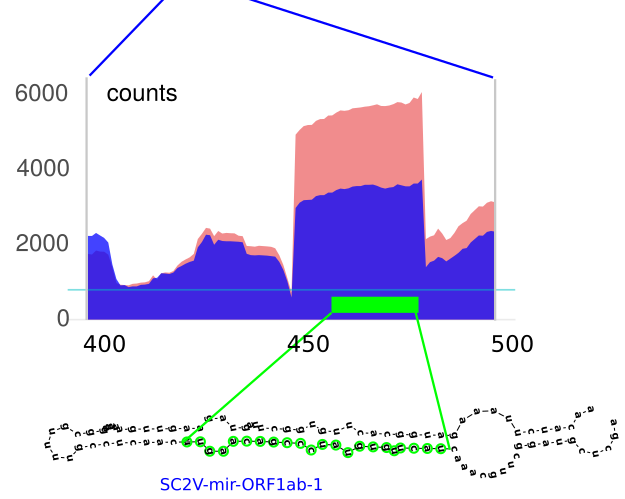 Novel SARS-CoV-2 encoded small RNAs in the passage to humansG. Merino, J. Raad, L. A. Bugnon, and 6 more authorsBioinformatics, May 2020
Novel SARS-CoV-2 encoded small RNAs in the passage to humansG. Merino, J. Raad, L. A. Bugnon, and 6 more authorsBioinformatics, May 2020The Severe Acute Respiratory Syndrome-Coronavirus 2 (SARS-CoV-2) has recently emerged as the responsible for the pandemic outbreak of the coronavirus disease 2019. This virus is closely related to coronaviruses infecting bats and Malayan pangolins, species suspected to be an intermediate host in the passage to humans. Several genomic mutations affecting viral proteins have been identified, contributing to the understanding of the recent animal-to-human transmission. However, the capacity of SARS-CoV-2 to encode functional putative microRNAs (miRNAs) remains largely unexplored. We have used deep learning to discover 12 candidate stem-loop structures hidden in the viral protein-coding genome. Among the precursors, the expression of eight mature miRNAs-like sequences was confirmed in small RNA-seq data from SARS-CoV-2 infected human cells. Predicted miRNAs are likely to target a subset of human genes of which 109 are transcriptionally deregulated upon infection. Remarkably, 28 of those genes potentially targeted by SARS-CoV-2 miRNAs are down-regulated in infected human cells. Interestingly, most of them have been related to respiratory diseases and viral infection, including several afflictions previously associated with SARS-CoV-1 and SARS-CoV-2. The comparison of SARS-CoV-2 pre-miRNA sequences with those from bat and pangolin coronaviruses suggests that single nucleotide mutations could have helped its progenitors jumping inter-species boundaries, allowing the gain of novel mature miRNAs targeting human mRNAs. Our results suggest that the recent acquisition of novel miRNAs-like sequences in the SARS-CoV-2 genome may have contributed to modulate the transcriptional reprograming of the new host upon infection. Available at https://github.com/sinc-lab/sarscov2-mirna-discovery
-
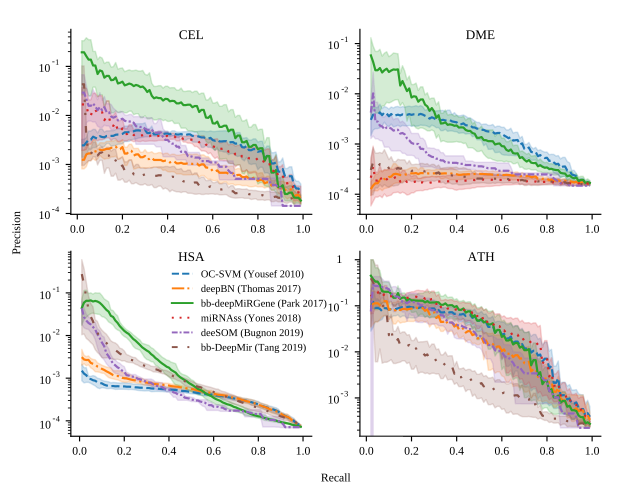 Genome-wide discovery of pre-miRNAs: comparison of recent approaches based on machine learningL. A. Bugnon, C. Yones, D. H. Milone, and 1 more authorBriefings in Bioinformatics, May 2020
Genome-wide discovery of pre-miRNAs: comparison of recent approaches based on machine learningL. A. Bugnon, C. Yones, D. H. Milone, and 1 more authorBriefings in Bioinformatics, May 2020Motivation The genome-wide discovery of microRNAs (miRNAs) involves identifying sequences having the highest chance of being a novel miRNA precursor (pre-miRNA), within all the possible sequences in a complete genome. The known pre-miRNAs are usually just a few in comparison to the millions of candidates that have to be analyzed. This is of particular interest in non-model species and recently sequenced genomes, where the challenge is to find potential pre-miRNAs only from the sequenced genome. The task is unfeasible without the help of computational methods, such as deep learning. However, it is still very difficult to find an accurate predictor, with a low false positive rate in this genome-wide context. Although there are many available tools, these have not been tested in realistic conditions, with sequences from whole genomes and the high class imbalance inherent to such data. Results In this work, we review six recent methods for tackling this problem with machine learning. We compare the models in five genome-wide datasets: Arabidopsis thaliana, Caenorhabditis elegans, Anopheles gambiae, Drosophila melanogaster, Homo sapiens. The models have been designed for the pre-miRNAs prediction task, where there is a class of interest that is significantly underrepresented (the known pre-miRNAs) with respect to a very large number of unlabeled samples. It was found that for the smaller genomes and smaller imbalances, all methods perform in a similar way. However, for larger datasets such as the H. sapiens genome, it was found that deep learning approaches using raw information from the sequences reached the best scores, achieving low numbers of false positives. Availability The source code to reproduce these results is in: http://sourceforge.net/projects/sourcesinc/files/gwmirna Additionally, the datasets are freely available in: https://sourceforge.net/projects/sourcesinc/files/mirdata
-
 DL4papers: a deep learning model for reading papersL. A. Bugnon, C. Yones, J. Raad, and 7 more authorsOxford Bioinformatics, May 2020
DL4papers: a deep learning model for reading papersL. A. Bugnon, C. Yones, J. Raad, and 7 more authorsOxford Bioinformatics, May 2020In precision medicine, next-generation sequencing and novel preclinical reports have led to an increasingly large amount of results, published in the scientific literature. However, identifying novel treatments or predicting a drug response in, for example, cancer patients, from the huge amount of papers available remains a laborious and challenging work. This task can be considered a text mining problem that requires reading a lot of academic documents for identifying a small set of papers describing specific relations between key terms. Due to the infeasibility of the manual curation of these relations, computational methods that can automatically identify them from the available literature are urgently needed. We present DL4papers, a new method based on deep learning that is capable of analyzing and interpreting papers in order to automatically extract relevant relations between specific keywords. DL4papers receives as input a query with the desired keywords, and it returns a ranked list of papers that contain meaningful associations between the keywords. The comparison against related methods showed that our proposal outperformed them in a cancer corpus. The reliability of the DL4papers output list was also measured, revealing that 100% of the first two documents retrieved for a particular search have relevant relations, in average. This shows that our model can guarantee that in the top-2 papers of the ranked list, the relation can be effectively found. Furthermore, the model is capable of highlighting, within each document, the specific fragments that have the associations of the input keywords. This can be very useful in order to pay attention only to the highlighted text, instead of reading the full paper. We believe that our proposal could be used as an accurate tool for rapidly identifying relationships between genes and their mutations, drug responses and treatments in the context of a certain disease. This new approach can certainly be a very useful and valuable resource for the advancement of the precision medicine field. A web-demo is available at: http://sinc.unl.edu.ar/web-demo/dl4papers/. Full source code and data are available at: https://sourceforge.net/projects/sourcesinc/files/dl4papers/.
2019
2019
-
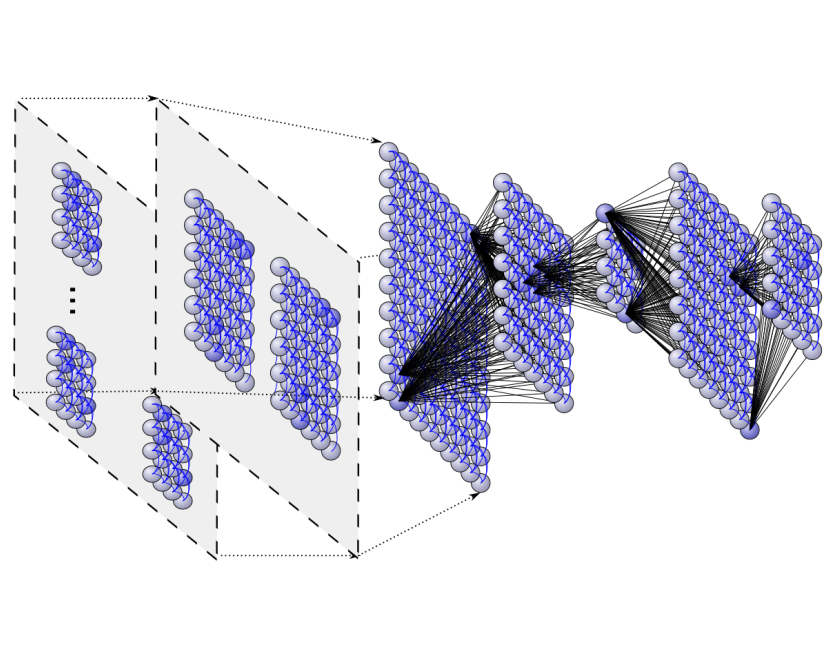 Deep neural architectures for highly imbalanced data in bioinformaticsL. A. Bugnon, Cristian A. Yones, D. H. Milone, and 1 more authorIEEE Transactions on Neural Networks and Learning Systems, May 2019
Deep neural architectures for highly imbalanced data in bioinformaticsL. A. Bugnon, Cristian A. Yones, D. H. Milone, and 1 more authorIEEE Transactions on Neural Networks and Learning Systems, May 2019In the postgenome era, many problems in bioinformatics have arisen due to the generation of large amounts of imbalanced data. In particular, the computational classification of precursor microRNA (pre-miRNA) involves a high imbalance in the classes. For this task, a classifier is trained to identify RNA sequences having the highest chance of being miRNA precursors. The big issue is that well-known pre-miRNAs are usually just a few in comparison to the hundreds of thousands of candidate sequences in a genome, which results in highly imbalanced data. This imbalance has a strong influence on most standard classifiers and, if not properly addressed, the classifier is not able to work properly in a real-life scenario. This work provides a comparative assessment of recent deep neural architectures for dealing with the large imbalanced data issue in the classification of pre-miRNAs. We present and analyze recent architectures in a benchmark framework with genomes of animals and plants, with increasing imbalance ratios up to 1:2000. We also propose a new graphical way for comparing classifiers performance in the context of high-class imbalance. The comparative results obtained show that, at a very high imbalance, deep belief neural networks can provide the best performance.
-
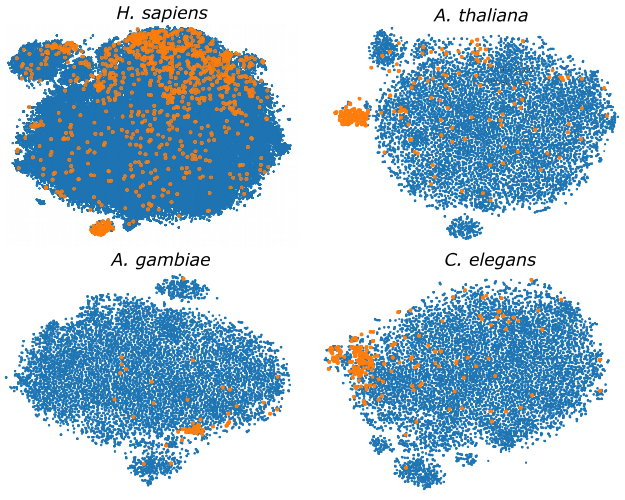 Genome-wide hairpins datasets of animals and plants for novel miRNA predictionL. A. Bugnon, C. Yones, D. H. Milone, and 1 more authorData in Brief, May 2019
Genome-wide hairpins datasets of animals and plants for novel miRNA predictionL. A. Bugnon, C. Yones, D. H. Milone, and 1 more authorData in Brief, May 2019This article makes available several genome-wide datasets, which can be used for training microRNA (miRNA) classifiers. The hairpin sequences available are from the genomes of: Homo sapiens, Arabidopsis thaliana, Anopheles gambiae, Caenorhabditis elegans and Drosophila melanogaster. Each dataset provides the genome data divided into sequences and a set of computed features for predictions. Each sequence has one label: i) “positive”: meaning that it is a well-known pre-miRNA, according to miRBase v21; or ii) “unlabeled”: indicating that the sequence has not (yet) a known function and could be a possible candidate to novel pre-miRNA. Due to the fact that selecting an informative feature set is very important for a good pre-miRNA classifier, a representative feature set with large discriminative power has been calculated and it is provided, as well, for each genome. This feature set contains typical information about sequence, topology and structure. Dataset was publically shared in https://sourceforge.net/projects/sourcesinc/files/mirdata/.
-
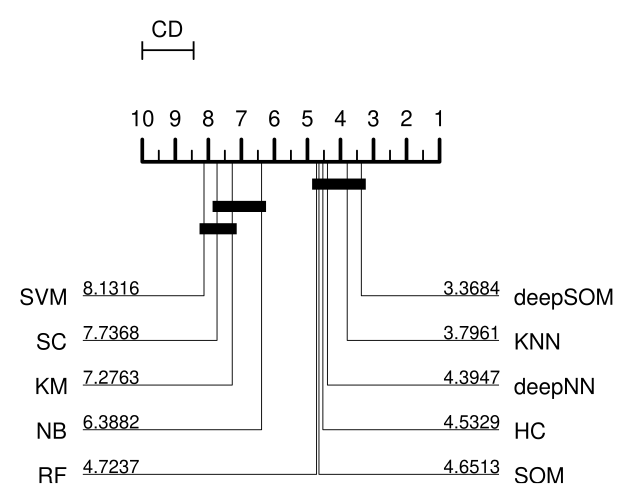 Predicting novel microRNA: a comprehensive comparison of machine learning approachesG. Stegmayer, L. Di Persia, Mariano Rubiolo, and 7 more authorsBriefings in Bioinformatics, May 2019
Predicting novel microRNA: a comprehensive comparison of machine learning approachesG. Stegmayer, L. Di Persia, Mariano Rubiolo, and 7 more authorsBriefings in Bioinformatics, May 2019The importance of microRNAs (miRNAs) is widely recognized in the community nowadays because these short segments of RNA can play several roles in almost all biological processes. The computational prediction of novel miRNAs involves training a classifier for identifying sequences having the highest chance of being precursors of miRNAs (pre-miRNAs). The big issue with this task is that well-known pre-miRNAs are usually few in comparison with the hundreds of thousands of candidate sequences in a genome, which results in high class imbalance. This imbalance has a strong influence on most standard classifiers, and if not properly addressed in the model and the experiments, not only performance reported can be completely unrealistic but also the classifier will not be able to work properly for pre-miRNA prediction. Besides, another important issue is that for most of the machine learning (ML) approaches already used (supervised methods), it is necessary to have both positive and negative examples. The selection of positive examples is straightforward (well-known pre-miRNAs). However, it is difficult to build a representative set of negative examples because they should be sequences with hairpin structure that do not contain a pre-miRNA. This review provides a comprehensive study and comparative assessment of methods from these two ML approaches for dealing with the prediction of novel pre-miRNAs: supervised and unsupervised training. We present and analyze the ML proposals that have appeared during the past 10 years in literature. They have been compared in several prediction tasks involving two model genomes and increasing imbalance levels. This work provides a review of existing ML approaches for pre-miRNA prediction and fair comparisons of the classifiers with same features and data sets, instead of just a revision of published software tools. The results and the discussion can help the community to select the most adequate bioinformatics approach according to the prediction task at hand. The comparative results obtained suggest that from low to mid-imbalance levels between classes, supervised methods can be the best. However, at very high imbalance levels, closer to real case scenarios, models including unsupervised and deep learning can provide better performance.
2017
2017
-
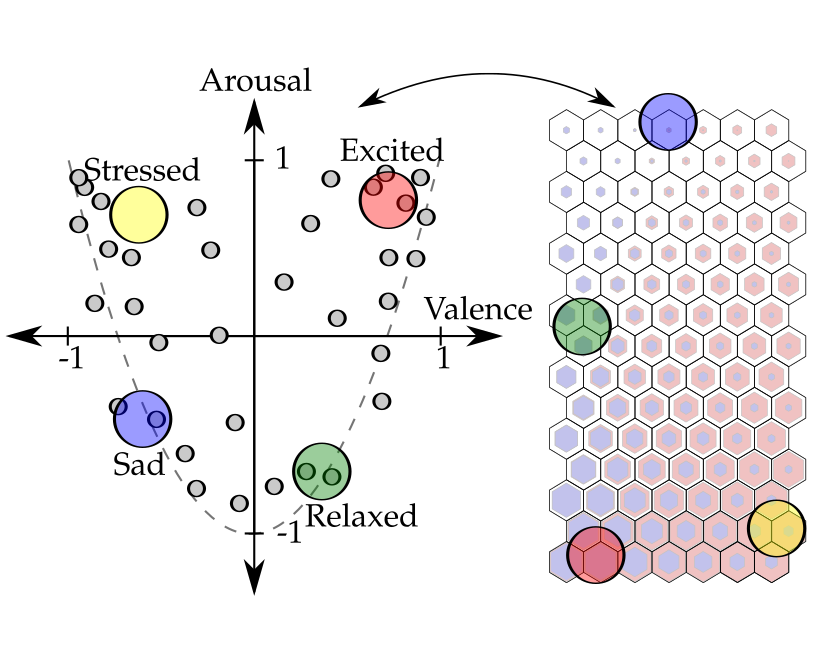 Dimensional Affect Recognition from HRV: an Approach Based on Supervised SOM and ELML. A. Bugnon, R. A. Calvo, and D. H. MiloneIEEE Transactions on Affective Computing, May 2017
Dimensional Affect Recognition from HRV: an Approach Based on Supervised SOM and ELML. A. Bugnon, R. A. Calvo, and D. H. MiloneIEEE Transactions on Affective Computing, May 2017Dimensional affect recognition is a challenging topic and current techniques do not yet provide the accuracy necessary for HCI applications. In this work we propose two new methods. The first is a novel self-organizing model that learns from similarity between features and affects. This method produces a graphical representation of the multidimensional data which may assist the expert analysis. The second method uses extreme learning machines, an emerging artificial neural network model. Aiming for minimum intrusiveness, we use only the heart rate variability, which can be recorded using a small set of sensors. The methods were validated with two datasets. The first is composed of 16 sessions with different participants and was used to evaluate the models in a classification task. The second one was the publicly available Remote Collaborative and Affective Interaction (RECOLA) dataset, which was used for dimensional affect estimation. The performance evaluation used the kappa score, unweighted average recall and the concordance correlation coefficient. The concordance coefficient on the RECOLA test partition was 0.421 in arousal and 0.321 in valence. Results show that our models outperform state-of-the-art models on the same data and provides new ways to analyze affective states.